Brief primer and lexicon for PacBio SMRT sequencing¶
PacBio SMRT sequencing operates within a silicon chip (a SMRTcell) fabricated to contain a large number of microscopic holes (ZMWs, or zero-mode waveguides), each assigned a hole number.
Within a ZMW, PacBio SMRT sequencing is performed on a circularized molecule called a SMRTbell. The SMRTbell, depicted below, consists of:
- the customer’s double-stranded DNA insert (with sequence \(I\), read following the arrow)
- (optional) double-stranded DNA barcodes (sequences \(B_L, B_R\)) used for multiplexing DNA samples. While the barcodes are optional, they must be present at both ends if present at all. Barcodes may or may not be symmetric, where symmetric means \(B_L = B_R^{RC}\).
- SMRTbell adapters (sequences \(A_L, A_R\)), each consisting of a double stranded stem and a single-stranded hairpin loop. Adapters may or may not be symmetric, where symmetric means \(A_L = A_R\).
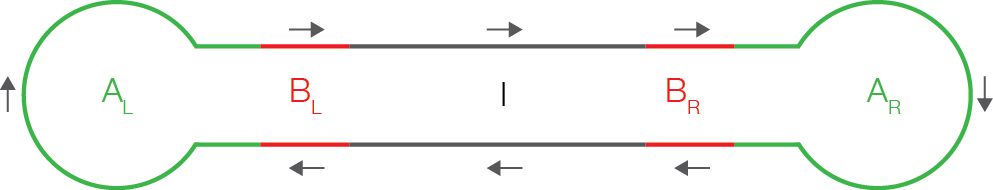
A schematic drawing of a SMRTbell
SMRT sequencing interrogates the incorporated bases in the product strand of a replication reaction. Assuming the sequencing of the template above began at START, the following sequence of bases would be incorporated (where we are using the superscripts C, R, and RC to denote sequence complementation, reversal, and reverse-complementation):
(note the identity \((x^{RC})^C = x^R\)).
The ZMW read is the full output of the instrument/basecaller upon observing this series of incorporations, subject to errors due to optical and other limitations. Adapter regions and barcode regions are the spans of the ZMW read corresponding to the adapter and barcode DNA. The subreads are the spans of the ZMW read corresponding to the DNA insert.
One complication arises when one considers the possibility that a ZMW might not contain a single sequencing reaction. Indeed it could could contain zero—in which case the ensuing basecalls are a product of background noise—or it could contain more than one, in which case the basecall sequence represents two intercalated reads, effectively appearing as noise. To remove such noisy sequence, the high quality (HQ) region finder in PostPrimary algorithmically detects a maximal interval of the ZMW read where it appears that a single sequencing reaction is taking place. This region is designated the HQ region, and in the standard mode of operation, PostPrimary will only output the subreads detected within the HQ region.
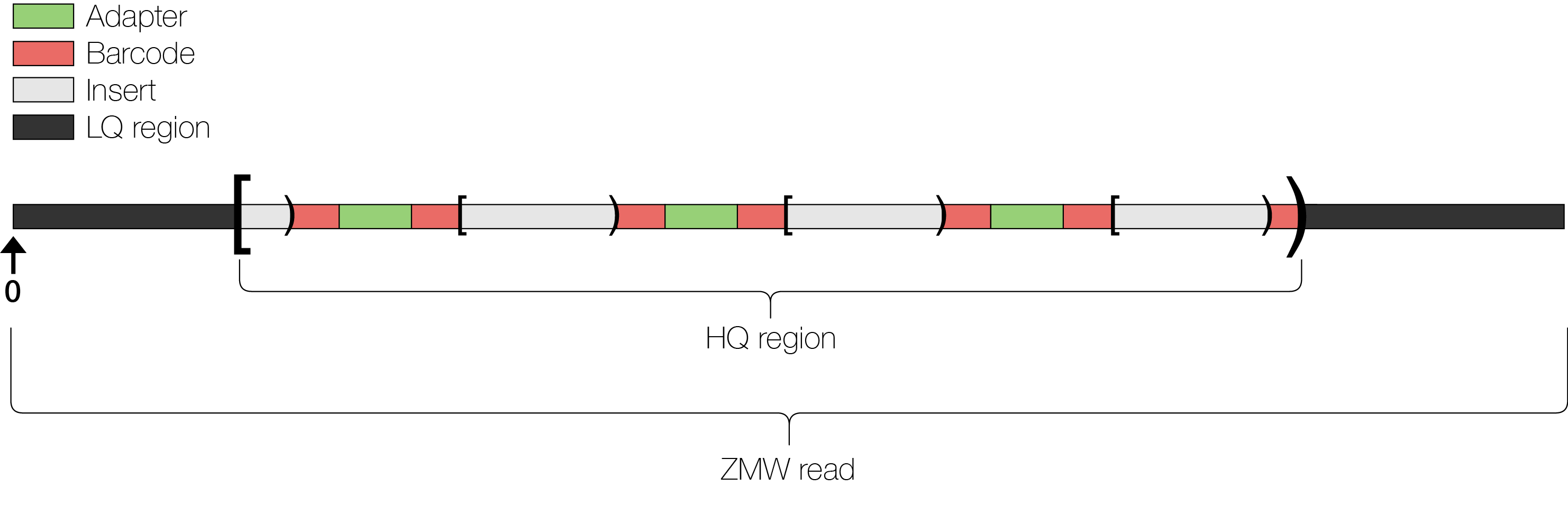
A schematic of the regions designated within a ZMW read
Note
Our coordinate system begins at the first basecall in the ZMW read (deemed base 0)—i.e., it is not relative to the HQ region. Intervals in PacBio reads are given in end-exclusive (“half-open”) coordinates. This style of coordinate system should be familiar to Python or C++ STL programmers.
BAM everywhere¶
Unaligned BAM files representing the subreads will be produced
natively by the PacBio instrument. The subreads BAM will be the
starting point for secondary analysis. In addition, the artifacts
resulting from removing adapter and LQ region sequences will be
retained in a scraps.bam file.
The circular consensus tool/workflow (CCS) will take as input an unaligned subreads BAM file and produce an output BAM file containing unaligned consensus reads.
Alignment (mapping) programs take these unaligned BAM files as input and will produce aligned BAM files, faithfully retaining all tags and headers.